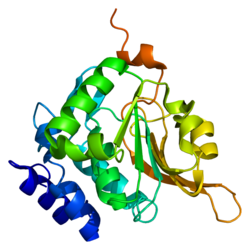
다중 서브 유닛 NADH:ubiquinone 산화환원효소(복합체 I)는 미토콘드리아의 전자전달 사슬에서 첫 번째 효소 복합체이다.철황단백질(IP) 분율은 NDUFS6를 [6]포함한 7개의 서브유닛으로 구성됩니다.
임상적 의의
NDUFS6 유전자의 돌연변이는 미토콘드리아 복합체 I 결핍과 관련이 있으며, 상염색체 열성 방식으로 유전된다.이 결핍은 산화적 인산화 [7][8]장애의 가장 흔한 효소적 결함이다.미토콘드리아 복합체 I 결핍은 극단적인 유전적 이질성을 나타내며 핵으로 인코딩된 유전자 또는 미토콘드리아로 인코딩된 유전자의 돌연변이에 의해 발생할 수 있다.명확한 유전자형-표현형 상관관계는 없으며 임상적 또는 생화학적 표현으로부터 기초적인 근거를 추론하는 것은 [9]불가능하지는 않더라도 어렵다.그러나 대부분의 경우 핵으로 인코딩된 [10][11]유전자의 돌연변이에 의해 발생한다.그것은 치명적인 신생아 질병에서 성인 신경 변성 장애까지 광범위한 임상 장애를 일으킨다.표현형에는 진행성 백혈구영양증, 비특이성 뇌증, 비대성 심근증, 근병증, 간질환, 리증후군, 르베르 유전성 시신경증, 파킨슨병 등이 포함된다.[12]
NDUFS6 돌연변이의 경우 일반적으로 다계통 장애와 [7][9][12]함께 빠르게 치명적으로 나타나는 신생아 젖산증입니다.
^"Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
^"Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
^Emahazion T, Beskow A, Gyllensten U, Brookes AJ (Nov 1998). "Intron based radiation hybrid mapping of 15 complex I genes of the human electron transport chain". Cytogenetics and Cell Genetics. 82 (1–2): 115–9. doi:10.1159/000015082. PMID9763677. S2CID46818955.
^McFarland R, Kirby DM, Fowler KJ, Ohtake A, Ryan MT, Amor DJ, Fletcher JM, Dixon JW, Collins FA, Turnbull DM, Taylor RW, Thorburn DR (Jan 2004). "De novo mutations in the mitochondrial ND3 gene as a cause of infantile mitochondrial encephalopathy and complex I deficiency". Annals of Neurology. 55 (1): 58–64. doi:10.1002/ana.10787. PMID14705112. S2CID21076359.
^Triepels RH, Van Den Heuvel LP, Trijbels JM, Smeitink JA (2001). "Respiratory chain complex I deficiency". American Journal of Medical Genetics. 106 (1): 37–45. doi:10.1002/ajmg.1397. PMID11579423.
Loeffen J, van den Heuvel L, Smeets R, Triepels R, Sengers R, Trijbels F, Smeitink J (Jun 1998). "cDNA sequence and chromosomal localization of the remaining three human nuclear encoded iron sulphur protein (IP) subunits of complex I: the human IP fraction is completed". Biochemical and Biophysical Research Communications. 247 (3): 751–8. doi:10.1006/bbrc.1998.8882. PMID9647766.
Loeffen JL, Triepels RH, van den Heuvel LP, Schuelke M, Buskens CA, Smeets RJ, Trijbels JM, Smeitink JA (Dec 1998). "cDNA of eight nuclear encoded subunits of NADH:ubiquinone oxidoreductase: human complex I cDNA characterization completed". Biochemical and Biophysical Research Communications. 253 (2): 415–22. doi:10.1006/bbrc.1998.9786. PMID9878551.
Brandenberger R, Wei H, Zhang S, Lei S, Murage J, Fisk GJ, Li Y, Xu C, Fang R, Guegler K, Rao MS, Mandalam R, Lebkowski J, Stanton LW (Jun 2004). "Transcriptome characterization elucidates signaling networks that control human ES cell growth and differentiation". Nature Biotechnology. 22 (6): 707–16. doi:10.1038/nbt971. PMID15146197. S2CID27764390.
Oh JH, Yang JO, Hahn Y, Kim MR, Byun SS, Jeon YJ, Kim JM, Song KS, Noh SM, Kim S, Yoo HS, Kim YS, Kim NS (Dec 2005). "Transcriptome analysis of human gastric cancer". Mammalian Genome. 16 (12): 942–54. doi:10.1007/s00335-005-0075-2. PMID16341674. S2CID69278.