직접 Xa 억제제 발견 및 개발
Discovery and development of direct Xa inhibitorsXa 직접 억제제 등급의 4가지 약이 전 세계적으로 시판되고 있다.리바록사반(샤렐토)은 2008년 유럽 및 캐나다에서 최초로 승인된 FXA 억제제였다.[1]두 번째는 2011년[2] 유럽, 2012년 미국에서 승인된 아픽사반(Eliquis)이다.[3]세 번째 에독사반(Lexiana, Savaysa)은 2011년 일본에서, 2015년 유럽과 미국에서 승인되었다.[4]베트릭사반(Bebyxa)은 2017년 미국에서 승인됐다.
역사
헤파린

헤파린은 1916년 제이 매클린과 윌리엄 헨리 하웰에 의해 발견되었는데, 이것은 그리스어로 헤파로 번역되는 개간에서 처음 격리되었다.헤파린은 혈액 응고 폭포에서 여러 인자를 대상으로 하는데 그 중 하나가 FXA이다.처음에는 많은 부작용이 있었지만, 그 후 20년 동안 조사관들은 헤파린을 더 좋고 안전하게 만들기 위해 연구했다.1935년에 임상시험에 들어갔고 1936년에 첫 약물이 출시되었다.천연 헤파린의 사슬은 5,000마리에서 40마일까지 다양하다.1980년대에 저분자 헤파린(LMWH)이 개발되었고 그것들은 평균 분자량이 8.000Da 미만인 체인만 포함하고 있다.[5]
와파린

1920년대에 캐나다와 미국 북부에서 의문의 출혈 소 질병이 발생했다.그 질병은 소들이 달콤한 클로버 건초를 풀을 뜯었기 때문에 달콤한 클로버 병이라고 이름 붙여졌다.현지 수사관 칼 P는 발병 후 10년이 지나서야 발병했다. 링크와 그의 제자 빌헬름 쇼에펠은 내출혈을 일으키는 물질을 찾기 위해 강도 높은 조사를 시작했다.그들이 원인 물질인 디코우마롤을 발견하는데 6년이 걸렸다.[5]그들은 그 물질에 대한 권리를 특허를 냈고 1945년에 링크는 설치류 살인으로 쿠마린 파생상품을 팔기 시작했다.그와 그의 동료들은 여러 가지 변형을 연구했고 결국 1948년에 와파린이라는 이름을 가진 물질을 갖게 되었다.1954년이 되어서야 와파린을 최초로 경구용 항응고제를 만드는 인간에게 약용으로 승인된 것이다.[6]
더 새롭고 더 나은 구강 약물의 필요성
와파린 치료는 치료 창이 좁아 정기적으로 혈액 모니터링과 투여량 조절이 필요하다.만약 감독이 적절하지 않다면 와파린은 너무 빈번하게 출혈을 일으키고 음식이나 다른 약물과 여러 번 상호작용을 일으키는데 위협을 가한다.현재 분자량 헤파린(LMWH)이 낮은 분자량 헤파린(Heparin, LMWH)의 주요 문제는 바로 행정경로인데, 이는 피하분자량(Heparin)을 투여해야 하기 때문이다.[7]이러한 단점들 때문에 더 나은 항응고제 약물이 절실히 필요하게 되었다.현대 사회에서, 편리하고 빠른 약물 투여는 좋은 약물 준수의 열쇠다.2008년에 최초의 직접 Xa 억제제가 임상 용도로 승인되었다.[8]다이렉트 Xa 억제제는 LMWH와 와파린만큼 효과적이지만 구두로 주어지며 엄격한 모니터링이 필요하지 않다.[7]다른 Xa 억제제 장점으로는 빠른 시작/오프셋, 약물 상호 작용이 거의 없고 예측 가능한 약동학 등이 있다.빠른 시작/오프셋 효과로 수술 후 자궁내항응고제와의 '브리지' 필요성이 크게 감소한다.[9]오늘날 시판되고 있는 Xa 억제제에는 4가지 요소가 있다: 리바록사반, 아픽사반, 에독사반, 베트릭사반.[7]
안티스타신 및 틱 항응고 펩타이드(TAP)
Factor Xa는 1980년대 초 새로운 항응고제 개발의 유망한 대상으로 확인되었다.1987년 제1인자, 자연적으로 발생하는 화합물인 안티스타신은 멕시코 해멘테리아 주례의 침샘으로부터 격리되었다.안티스타신은 폴리펩타이드와 강력한 Xa 억제제다.1990년에 또 다른 자연발생적인 Xa 억제제는 Ornithodoros moubata 추출물에서 틱 항응고 펩타이드(TAP)를 분리했다.TAP와 안티스타신은 인자 Xa를 약물 표적으로 추정하기 위해 사용되었다.[8]
작용기전

혈액 응고는 혈액이 응고를 형성하는 복잡한 과정이다.지혈의 필수적인 부분이며 손상된 혈관의 출혈을 막음으로써 작용한다.[10]내피 아래 피에 노출되는 부상 부위에 혈소판이 모여 곧바로 플러그를 형성한다.그 과정은 1차 지혈이라고 불린다.동시에 2차 지혈이 일어난다.그것은 특히 트롬빈에 의한 활성 응고 인자에 의한 불용성 피브린의 형성으로 정의된다.[11]이러한 요인들은 내적 경로와 외적 경로인 두 개의 분리된 경로를 통해 발생하는 혈액 응고 폭포에서 서로를 활성화시킨다.[12]다양한 프로엔자임을 활성화한 뒤 계단식 마지막 단계에서 트롬빈이 형성돼 피브리노겐을 피브린으로 변환해 응고 형성을 유도한다.[10]Factor Xa는 활성 세린 프로테아제로, 프로트롬빈을 트롬빈으로 변환하여 혈액 응고 경로의 핵심 역할을 차지한다.Xa 인자의 억제는 트롬빈의 양을 줄임으로써 항혈전 효과로 이어진다.Xa를 직접 목표로 하는 요소는 항응고 작용에 대한 효과적인 접근법이라고 제안된다.[8]
개발

1987년 안티스타신은 최초의 직접 Xa 억제제로 시험되었다.안티스타신은 119개의 아미노산 잔류물로 구성된 단백질로 이황화합물 10개에 관여하는 시스테인이 20개다.[13]0.3~0.6nM의 Ki 값을 갖는 인자 Xa의 느리고 촘촘한 구속 억제제 역할을 하지만 트립신을 억제하기도 한다.[8]안티스타신 재조합은 유전적으로 조작된 효모, 사카로마이오스 세레비시아에 의해 만들어질 수 있다.[14]또 다른 자연발생적인 Xa-inhibitor인 진드기 항응고 펩타이드(TAP)는 1990년에 발견되었다.단일 체인 60 아미노산 펩타이드로 안티스타신처럼 Ki 값(~0.6nM)이 비슷한 느리고 촘촘한 결합 억제제다.
이 두 가지 단백질은 주로 Xa라는 인자를 약물 표적으로 검증하는 데 사용되었다.동물 연구는 직접적 Xa-inhibition이 직접 혈청 억제제에 비해 항응고에 대한 보다 효율적인 접근방법이 될 것을 제안했으며, 특히 d에 비해 치료 창을 넓히고 반발 혈전증 위험을 줄였다(항혈전증 치료제 철회 직후에 발생하는 혈전증 사건의 증가).이직 및 간접 트롬빈 억제제.[8]
1990년대에 DX-9065a와[15] YM-60828과 같은 몇몇 저분자-중량 물질이 개발되었다.[16]
_and_DX-9065a_(right).jpg)
DX-9065a는 트롬빈을 억제하지 않고 FXa를 억제한 최초의 합성 화합물이었다.그것은 FXA에 대한 선택적 결합에 가장 중요한 계류인 것 같은 카복실 그룹을 삽입함으로써 얻어졌다.[8]초기에 개발된 작은 분자들은 아직 아미딘 그룹이나 심지어 더 높은 기본적 기능을 가지고 있었는데, 이것은 인자 Xa의 자연 기질인 프로트롬빈에서 아르기닌 잔여물의 모사로서 필요하다고 생각되었다.그럼에도 불구하고, 이러한 기본적인 기능들은 또한 매우 낮은 구강 생체이용률과 관련이 있다(예: DX-9065a의 경우 2~3%).
1998년 한 제약회사가 경구 생체이용률이 높은 저분자중량 직접인자 Xa 억제제를 찾기 시작했다.처음에는 고투과 스크리닝과 추가 최적화가 이소인돌리논 등급의 여러 물질로 이어져 훨씬 더 적은 기본 물질이 최대 2nM의 IC50 값에 대한 강력한 Xa 억제제 역할도 할 수 있음을 입증한다.비록 이소인돌리논이 원래의 화합물보다 더 나은 경구 생체이용률을 가지고 있지만 충분하지 않았다.그러나, 이 프로젝트는 후에 인자인 Xa를 억제하는 높은 효력과 높은 생체이용성을 모두 가진 물질을 제공하는 n-aryloxazolidones의 등급으로 이어진다.[8]이 세분류의 한 화합물인 리바록사반(IC50 = 0.7nM, 생체이용률: 60%)은 2008년 9월 유럽과 캐나다에서 정맥 혈전증 예방을 위한 마케팅 허가를 받았다.[1][17]
화학
요인 Xa: 구조 및 바인딩 사이트

인자 IIa, Xa, VIIa, IXa, XIa는 모두 응고 폭포에서 특정한 역할을 하는 단백질 분해 효소다.인자 Xa(FXa)는 내인 경로와 외인 경로의 교차점에서의 위치뿐만 아니라 각 Xa 분자에 대해 약 1000 트롬빈 분자를 생성하여 강력한 항응고 효과를 초래하기 때문에 가장 유망하다.FXa는 인자 Xa의 "a"가 활성화됨을 의미하듯이 52개의 아미노산 활성화 펩타이드의 분해에 의해 FX에서 생성된다.FXa는 254개의 아미노산 촉매 영역으로 구성되며 142개의 아미노산 조명 체인과도 연결된다.체인은 GLA 도메인과 두 개의 표피 성장 인자 도메인(EGF like domain)을 모두 포함한다.[18]
FXa의 활성 부위는 생리적 기판의 갈라진 틈을 촉매로 만들어 프로트롬빈에 있는 PhePePeAsnProArg-ThrPho와 TyrIleAspG-IleVal을 클리핑하도록 구성되었다.FXa는 Xa 인자에 결합할 기판들의 표적이 되는 소위 4개의 포켓을 가지고 있다.이 주머니들은 다른 아미노산에 의해 정렬되어 있고 Xa 억제제는 Xa 인자에 결합할 때 이 주머니들을 목표로 한다.Xa 억제제에 대한 친화력과 선택성과 관련하여 가장 관련성이 높은 두 개의 포켓은 S1과 S4이다.[18]
S1: S1 포켓은 소수성 포켓이며 아스파르트산 잔류물(Asp-189)을 함유하고 있어 기본 집단의 인식장소 역할을 할 수 있다.FXa는 S1 포켓에 잔여 공간이 있으며 잔류물 Tyr-228, Asp-189 및 Ser-195로 정렬되어 있다.[18]
S2: S2 포켓은 작고 얕은 주머니다.S4 포켓과 합쳐져 작은 아미노산이 들어갈 수 있는 공간이 있다.Tyr-99는 이 주머니의 접근을 막는 것 같아 이 주머니는 S1이나 S4만큼 중요하지 않다.[19]
S3: S3 포켓은 S1 포켓의 테두리에 위치하며 납작하여 용제에 노출된다.이 주머니는 S1이나 S4만큼 중요하지 않다.
S4: S4 포켓은 자연적으로 소수성이며 주머니 바닥은 Trp-215 잔여물로 형성된다.잔류물인 FXA의 Peh-174와 Tyr-99는 Trp-215와 결합해 발광성, 방향성, 양전하 파편을 결합할 수 있는 방향성 박스를 형성한다.양전하 기업에 대한 구속력 때문에, 그것은 양이온 구멍이라고 말할 수 있다.[18]
직접자아 억제제의 화학구조 및 특성
| 리바록사반 | 아픽사반 | 에독사반 | |
|---|---|---|---|
| MW(g/mol) | 436 | 460 | 548 |
| 분자식 | CHClNos | C25H25N5O4 | CHClNos |
| 모양 | L | L | L |
| Ki | 0.4nM | 0.08nM | 0.561nM |
| IC50 | 0.7nM | 해당 없음 | 해당 없음 |
| 경구 생체이용률(%) | 66–100 (1969년) | 50 | 62 |

Xa 억제제를 인자 Xa에 결합
Xa 억제는 모두 인자 Xa의 활성 사이트 내에서 소위 L자형 패션으로 결합한다.Xa 인수의 핵심 구성 요소는 S1과 S4 바인딩 사이트다.극성이 높고 따라서 충전된 구성 요소를 가진 자연 화합물인 안티스타신 및 TAP가 어느 정도 특정성을 가지고 목표물에 결합한다는 것이 처음 주목되었다.그래서 신약들은 긍정적으로 충전된 그룹들로 설계되었지만 그것들 때문에 생체이용성이 떨어지는 결과를 낳았다.오늘날 시판되고 있는 Xa 억제제는 따라서 S1 및 S4 결합 사이트와의 다양한 상호작용을 위해 다양한 모이티(moieties)가 부착된 방향족 고리를 포함한다.이것은 또한 강력한 결합 강도를 유지하는 것뿐만 아니라 좋은 생체 이용가능성을 보장한다.현재 시판 중인 Xa 억제제는 극성 상호작용 대신 소수성과 수소 결합에 의존한다.[20]
인자 Xa에 대한 안티스타신 바인딩
안티스타신은 아미노산 서열에서 유사한 N-와 C-단자영역을 포함하고 있으며, 아미노산 서열은 약 40%, 호몰로지 약 56%로 유사하다.각각 짧은 β-시트 구조와 이황화물 결합 5개를 포함하고 있다.C-단자 영역은 실제 활성 사이트에 강한 아날로그 패턴을 가지고 있음에도 불구하고, C-단자 영역은 3차원 구조의 차이로 인한 억제 특성에 기여하지 않는 반면 Xa를 억제하기 위해서는 N-단자 영역만 필요하다.[13]
안티스타신과 FXa의 상호작용은 FXa의 활성 부위와 비활성 표면을 모두 포함한다.N-terminal 도메인에서 Arg-34와 Val-35에 의해 형성된 안티스타신의 반응성 부지는 FXa의 바인딩 부지에 적합하며, 아마도 S1 포켓일 것이다.동시에 안티스타신 반응 부위 외부에 위치한 Glu-15는 FXa 표면에 양전하 잔류물에 적합하다.다중 결합은 열역학적으로 유리하며 나노극 이하 억제(Ki = 0.3–0.6nM[8])로 이어진다.[13]
DX-9065a 인자 Xa 바인딩
DX-9065a는 최초의 소형 분자 직접 Xa-inhibitor로 분자량이 571.07g/mol인 아미디노아릴 유도체다.[21]양극으로 충전된 아미노나프탈렌 그룹은 FXA의 S1 포켓에 있는 아스프-189 잔여물에 대한 염교 역할을 한다.피롤리딘 링은 FXA의 S4 포켓에 있는 Tyr-99, Phe-174, Trp-215 사이에 맞는다.[22]
헤파린 등 기존 약물과 달리 FX-9065a는 구조상 FXa와 트롬빈이 유사하지만 트롬빈에 비해 FXa가 선택적이다.이는 호몰로뉴 위치 192에서 아미노산 잔류물의 차이로 인해 발생한다.FXa는 글루타민 잔류물이 그 위치에 있는 반면, 트롬빈은 글루탐산을 가지고 있어 DX-9065a의 카르복실 그룹과 정전기적 저항을 일으킨다.또한 트롬빈의 글루-97과 DX-9065a의 피롤리딘 링에 고정된 아미딘 그룹 사이의 염교는 DX-9065a 분자의 유연성을 감소시켜 이제 정전기 충돌을 피하기 위해 충분히 회전할 수 없게 되었다.그렇기 때문에 트롬빈의 IC50 값은 1000µM 이하인 반면 FXa의 IC50 값은 0,16µM이다.[22]

인자에 대한 리바록사반 결합
FXA에 대한 리바록사반 결합은 아미노산 글리-219에 두 개의 수소 결합을 통해 매개된다.이 두 개의 수소 결합은 이 약을 FXA의 S1과 S4 서브사이트로 유도하는 중요한 역할을 한다.첫 번째 수소 결합은 리바록사반의 옥사졸리디논 핵의 카보닐 산소로부터 오는 강한 상호작용이다.두 번째 수소 결합은 더 약한 상호작용이며 클로로티오페네 카르박스아미드 몰리의 아미노 그룹에서 나온다.
이 두 개의 수소 결합으로 인해 약물이 L자형을 형성하게 되고 S1과 S4 포켓에 들어맞는다.아미노산 잔류물인 Phe-174, Tyr-99, Trp-215는 S4 바인딩 포켓인 좁은 소수성 채널을 형성한다.리바록사반의 모폴리논 부분은 아미노산 Tyr-99와 Phe-174 사이에 "샌드위치드"되며, 리바록사반의 아릴 링은 Trp-215를 가로지르는 수직 방향이다.모폴리논 카보닐 그룹은 FXA 백본과 직접적인 상호작용을 하지 않고 대신, 모폴리논 링의 평면화에 기여하므로 리바록사반이 두 아미노산 사이에 끼도록 지원한다.
티오페인 계통의 염소 대체물과 S1의 하단에 위치한 Tyr-228의 방향족 링의 상호작용은 FXa에 대한 높은 친화력을 위해 강한 기초 집단의 필요성을 없애주기 때문에 매우 중요하다.이를 통해 비기초적인 리바록사반이 좋은 구강 생체이용률과 효용성을 달성할 수 있다.[8]

아픽사반 결합 인자 Xa
아픽사반은 리바록사반과 유사한 결합 모드를 보이며, FXA에 연결되면 엄격한 억제제-엔자임 콤플렉스를 형성한다.아픽사반의 p-methoxy 그룹은 FXa의 S1 포켓에 연결되지만, 이 FXa 지역의 잔류물과 어떠한 상호작용도 하지 않는 것으로 보인다.아픽사반의 피라졸 N-2 질소 원자는 Gln-192와 상호작용하고 카보닐 산소는 글리-216과 상호작용한다.아픽사반의 페닐 락탐 그룹은 Tyr-99와 Phe-174 사이에 위치하며 그 방향성 때문에 S4 포켓의 Trp-215와 상호작용할 수 있다.락탐모이성의 카보닐 산소 그룹은 물 분자와 상호작용을 하며 S4 포켓의 어떤 잔류물과도 상호작용을 하지 않는 것으로 보인다.[23]

구조-활동-관계(SAR)
특정 대상에 대한 이상적인 억제제인 화합물 설계의 중요한 부분은 화합물이 결합할 대상 부지의 아미노산 순서를 이해하는 것이다.프로트롬빈과 에프엑스아 모두를 모델링하면 그 차이를 차감하고 각 결합 부위에서 아미노산을 식별할 수 있다.FXa의 S1 포켓 하단에서 결합 아미노산은 아미딘 모이에티가 결합할 수 있는 아스프-189이다.FXa의 바인딩 부지를 X레이로 촬영한 결과 S1 포켓은 평면형상이 있어 평평한 아미디노아릴 집단이 장력 방해 없이 바인딩해야 한다는 의미가 있는 것으로 밝혀졌다.[8]
현대의 직접 Xa 억제제는 끝이 S1과 S4 포켓에 완벽하게 맞는 L자형 분자다.L자형의 긴 면은 대상 활성 사이트 내의 매우 특정한 터널에 따라야 한다.이를 위해 분자의 이 부분은 그 부위의 FXA와 거의 공식적 상호작용을 하지 않도록 설계되었다.특정한 결합이 없기 때문에, FXa의 주머니 사이에 이러한 작용제를 장착하면 FXa 분자에 대한 약물의 전체 특수성이 증가한다.FXa의 S1 포켓과 억제제 사이의 상호작용은 이온성 또는 비이온성이 될 수 있는데, 이는 모이에티 설계를 조정하여 경구 생체이용률을 높일 수 있도록 하기 때문에 중요하다.이전에 고안된 화합물은 위장관에 잘 흡수되지 않아 높은 혈청 농도에 도달하지 못하는 충전된 분자였다.신약은 충전되지 않고 S1 포켓과 비이온적 상호작용을 하기 때문에 생체이용성이 더 좋다.[20]
리바록사반
리바록사반의 SAR 개발 중에, 연구자들은 옥사졸리도닌 코어에 5-클로로티오페네-2-카복사미드 그룹을 첨가하면 이전에는 의료용으로 너무 약했던 효력이 200배 증가할 수 있다는 것을 깨달았다.이 발견 외에도 (S)-구성에 대한 명확한 선호도가 확인되었다.이 화합물은 유망한 약동학 프로파일을 가지고 있었고 매우 기본적인 아미딘 그룹을 포함하고 있지는 않았지만, 이전에는 S1 포켓과의 상호작용을 위해 중요한 것으로 여겨져 왔다.이러한 연구 결과는 광범위한 SAR(구조-활동 관계) 연구로 이어진다.SAR 시험 동안, R1은 효력에 대한 가장 중요한 그룹으로 정의되었다.Pyrrolidinone은 R1 기능 그룹 중 최초로 효력을 크게 증가시켰지만, 추가 연구에서는 모폴리논 그룹보다 훨씬 더 높은 효력이 밝혀졌다.그룹 R2와 R3에는 수소나 불소가 부착돼 있었고 수소를 가졌을 경우 효력이 가장 높은 것으로 빠르게 평가됐다.이후 그룹 R2와 R3가 수소보다 모두 위력이 떨어지는 다양한 그룹을 대체하게 되었기 때문에 수소가 최종 결과였다.클로로티오페네 해열은 용해도가 부적절해 이를 다른 집단으로 대체하려 했으나 성공하지 못했다.클로로티오페네 모이티는 S1 포켓 하단의 Tyr-228에 결합되어 FXA와의 결합에 관한 핵심 요소가 된다.리바록사반은 친화력이 높고 생체이용률이 좋다.[24]

아픽사반

아픽사반의 SAR 개발 동안 최대 효용성과 생체이용성을 얻기 위해 테스트가 필요한 세 개의 그룹이 있었다.p-methoxyphenyl 그룹(S1 바인딩 moiety)에 대한 SAR 테스트 전에 안정화해야 하기 때문에 첫 번째로 테스트한 그룹은 비활성 사이트였다.대부분 아미드, 아민, 테트라졸 등 화합물의 효능을 높이는 여러 집단이 있지만 메틸설포닐, 트라이플루오로메틸 그룹도 있다.이 그룹 중 카르복사미드는 결합력이 가장 크고 화합물과 유사한 응고 활성을 가지고 있다.[25]
개 실험에서, 카복사미드 그룹인 13F와 함께 이 화합물은 뛰어난 약동학적 프로파일과 낮은 간극, 적절한 반감기와 분포를 보였다.안정화 그룹 발굴의 성공으로 S1 바인딩 모이에티(p-메톡시페닐)에 대한 SAR 연구가 중단됐다.S4 바인딩 그룹에서 N-메틸아세틸과 락탐 유사 물질은 FXa에 대한 결합 친화력이 매우 높은 것으로 입증되었으며, 다른 프로테아제 대비 높은 응고와 선택성을 보였다.아세타미드 대비 N-메틸 아세틸의 FXA 결합 능력이 S4 지역 결합 부지에 가까운 불리한 평면성으로 인해 300배 낮았기 때문에 오리엔테이션이 중요한 것으로 나타났다.[25]
합성
리바록사반
리바록사반은 화학적으로 n-aryloxazolidones 그룹에 속한다.그 그룹의 다른 약들은 라인졸리드, 테디졸리드인데 둘 다 항생제다.오실릴 보호 에틸(2,3-디하이드록시프로필)-카르바메이트로 시작하는 n-아릴록사졸리디논 합성이 2016년 출간됐다.1배트의 반응으로 카바메이트 사이클은 약간 기본적인 조건에서 2-옥사졸리돈 링으로 가는 반면, 동시에 옥사졸리돈 질소는 구리 촉매화에 의해 아릴화된다.특히 리바록사반의 경우 3-모폴롤리논이 벤젠 링의 p-위치에 있는 요오드를 구리-소산화에 의해 대체한다.그 후, 실릴 보호 그룹은 제거되고 그 결과 알코올은 아미노 그룹으로 대체되며, 아미노 그룹은 마지막 단계에서 아틸화된다.[26]
리바록사반의 산업 준비는 바이엘헬스케어가 2005년 특허로 등록했다.[27]프탈리미드 보호 그룹에 관여하는 1차 아민이 함유된 프로필렌 산화물 유도체에 의해 알킬화 된 N-(4-aminophenol)-모르폴리논에서 시작한다.다음으로 포스겐 등가물이 첨가되어 2-옥사졸리돈 링을 형성하고 프탈리미드를 제거한다.프리 아민은 이제 아세틸화 되어 리바록사반으로 이어질 수 있다.
그러나, 특허에 따르면, 합성은 "준비하기 위해 특히 불리한 영향을 미치는 반응 관리에서 다양한 불이익"을 가지고 있다.특허는 또한 산업 공정에 더 적합할 클로로티오페인 유도체로부터 시작하는 또 다른 합성을 설명하지만 독성 용제나 시약을 최종 제품에서 제거해야 한다고 지적한다.그러므로 이런 방법은 대안이 아니다.[27]
리바록사반의 다양한 다른 합성경로가 설명되어 왔다.[28][29]
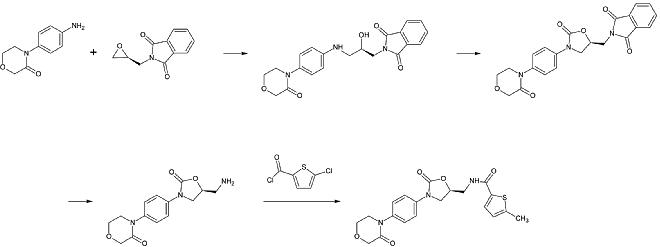
첫 번째 단계:1차 방향족 알킬레이션
두 번째 단계:포스겐 등가물을 이용한 2-옥사졸리돈 링 형성
세 번째 단계:프탈리미드 보호군 제거
네 번째 단계:1차 아민의 아킬레이션
아픽사반
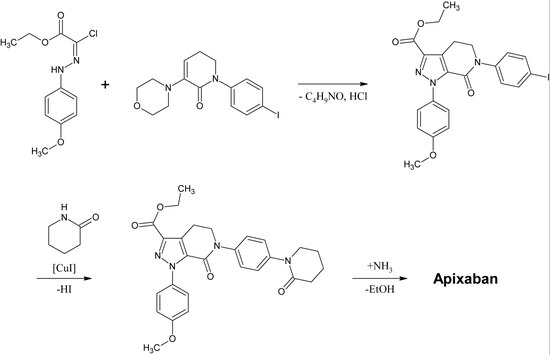
아픽사반의 최초 완전합성은 2007년에 출판되었다.[30]이 반응의 핵심 단계는 p-methoxyphenyl cloridrazon 유도체와 p-iodophenyl-모르폴린-dihydropyridin 유도체의 (3+2) 사이클로어다.다음과 같이 HCl과 모폴린을 제거한 후, 요오드는 구리-소산화에 의해 2-피페리디논으로 대체되고 에틸에스더는 아미드(아미노리시스)로 변환된다.이 반응은 2009년 특허로 등록됐다.[31]
임상용도
직접 인자 Xa 억제제는 임상적으로 사용되고 있으며 그 사용은 지속적으로 증가하고 있다.와파린 사용량과 낮은 분자량 헤파린(LMWH)을 점차 인수하고 있다.[8]Xa 억제제 표시가 폐색전증을 유발할 수 있는 심부정맥혈전증(DVT)을 예방하고 있다.또한 심방세동 치료에도 사용되어 혈전으로 인한 뇌졸중 위험을 낮춘다.또 다른 징후는 동맥경화로 인한 혈액 응고(혈전증)에 대한 예방적 치료법이다.리바록사반(Rivaroxaban)은 시판된 최초의 FXA 억제제였으며, 아픽사반(apixaban), 에독사반(edoxaban), 베트릭사반(betrixaban)이 그 뒤를 이었다.
| 리바록사반 | 아픽사반 | 에독사반 | 베트릭사반 | |
|---|---|---|---|---|
| 브랜드명 | 사렐토 | 엘리키스 | 사바이사, 리시아나 | 베빅사 |
| 개발자 및 생산자 | 바이어 | 화이저 | 다이이치상쿄 | 포톨라 제약 |
약동학
| 리바록사반 | 아픽사반 | 에독사반 | |
|---|---|---|---|
| 신진대사 | CYP3A4/5(주요), CYP2J2(소형) | CYP3A4(주요), CYP1A2, 2C8, 2C19, 2J2(모두 마이너) | CYP34A(주요) |
| 단백질 결합(%) | 92–95 | 87 | 55 |
| 반감기(hrs) | 5–9 | 6–12 | 5–11 |
| 제거 | 레날 (66%; 불변 약물로 36% 36%) | 레날(27%), 대변 | 레날(35%) |
| 흡수(Tmax) | 2~4시간 | 3~4시간 | 1~2시간 |
| 분포(L) | 50 | 21–61 | 107 |
| 신장 간극(L/hr) | 2.4 | 7.5 | 11 |
미래 관점
임상 실험에서 직접 Xa 억제제
리바록사반, 아픽사반, 에독사반, 베트릭사반이 이미 시중에 나와 있다.2016년 10월 현재 몇 가지 새로운 Xa 직접 억제제가 임상시험에 들어갔다.다케다에서 온 레탁사반과 화이저에서 온 에리박사반이다.[34]
해독제
포톨라제약의 안덱사(Andexanet alfa)는 정맥주입되는 재조합성 단백질이다.그것은 모든 직간접적인 FXa 억제제의 해독제 역할을 한다.안덱사는 Xa 억제제의 데코이 수용체 역할을 한다.
참조
- ^ a b "European Medicines Agency. 2016. Xarelto". www.ema.europa.eu. Retrieved 2016-10-03.
- ^ "European Medicines Agency. 2016. Eliquis". www.ema.europa.eu. Retrieved 2016-10-03.
- ^ Bhanwra, Sangeeta; Ahluwalia, Kaza (2014-01-01). "The new factor Xa inhibitor: Apixaban". Journal of Pharmacology and Pharmacotherapeutics. 5 (1): 12–4. doi:10.4103/0976-500x.124409. PMC 3917159. PMID 24554904.
- ^ Chan, Lewey; Pisano, Michele (2016-10-03). "Edoxaban (Savaysa): A Factor Xa Inhibitor". Pharmacy and Therapeutics. 40 (10): 651–95. ISSN 1052-1372. PMC 4606855. PMID 26535021.
- ^ a b Wardrop, D.; Keeling, D. (2008). "The story of the discovery of heparin and warfarin". British Journal of Haematology. 141 (6): 757–63. doi:10.1111/j.1365-2141.2008.07119.x. PMID 18355382.
- ^ Francis, CW. (2008). "Warfarin: An Historical Perspective". Hematology. 2008: 251. doi:10.1182/asheducation-2008.1.251. PMID 19074091.
- ^ a b c Massimo, F.; Mannucci, P.M (2016). "Direct oral anticoagulants and venous thromboembolism". European Respiratory Review. 25 (141): 295–302. doi:10.1183/16000617.0025-2016. PMID 27581829.
- ^ a b c d e f g h i j k Perzborn, E.; Roehrig, S.; Straub, A.; Kubitza, D.; Misselwitz, F. (2011). "The discovery and development of rivaroxaban, an oral, direct factor Xa inhibitor". Nature Reviews Drug Discovery. 10 (1): 61–75. doi:10.1038/nrd3185. PMID 21164526.
- ^ Bauer, K.A. (2013). "Pros and cons of new oral anticoauglants". Hematology. 2013: 464–70. doi:10.1182/asheducation-2013.1.464. PMID 24319220.
- ^ a b Furie, B; Furie, B.C. (2008). "Mechanisms of thrombus formation". The New England Journal of Medicine. 359 (9): 938–49. doi:10.1056/nejmra0801082. PMID 18753650.
- ^ Davie, E.W.; Fujikawa, K; Kisiel, W (1991). "The coagulation cascade: initiation, maintenance, and regulation". Biochemistry. 30 (43): 10363–70. doi:10.1021/bi00107a001. PMID 1931959.
- ^ Mackman, N; Tilley, R.E.; Key, N.S. (2007). "Role of the Extrinsic Pathway of Blood Coagulation in Hemostasis and Thrombosis". Arteriosclerosis, Thrombosis, and Vascular Biology. 27 (8): 1687–93. doi:10.1161/atvbaha.107.141911. PMID 17556654.
- ^ a b c Lapatto, R.; Krengel, U.; Schreuder, H. A.; Arkema, A.; de Boer, B.; Kalk, K. H.; Hol, W. G.; Grootenhuis, P. D.; Mulders, J. W. (1997-09-01). "X-ray structure of antistasin at 1.9 A resolution and its modelled complex with blood coagulation factor Xa". The EMBO Journal. 16 (17): 5151–61. doi:10.1093/emboj/16.17.5151. ISSN 0261-4189. PMC 1170148. PMID 9311976.
- ^ Schultz, Loren D.; Markus, Henry Z.; Hofmann, Kathryn J.; Montgomery, Donna L.; Dunwiddie, Christopher T.; Kniskern, Peter J.; Freedman, Robert B.; Ellis, Ronald W.; Tuite, Michael F. (1994-06-01). "Using Molecular Genetics to Improve the Production of Recombinant Proteins by the Yeast Saccharomyces cerevisiae". Annals of the New York Academy of Sciences. 721 (1): 148–57. doi:10.1111/j.1749-6632.1994.tb47387.x. ISSN 1749-6632. PMID 8010665.
- ^ Nagahara, Takayasu; Yokoyama, Yukio; Inamura, Kazue; Katakura, Shin-ichi; Komoriya, Satoshi; Yamaguchi, Hitoshi; Hara, Tsuyoshi; Iwamoto, Masahiro (1994-04-01). "Dibasic (Amidinoaryl)propanoic Acid Derivatives as Novel Blood Coagulation Factor Xa Inhibitors". Journal of Medicinal Chemistry. 37 (8): 1200–07. doi:10.1021/jm00034a018. ISSN 0022-2623. PMID 8164262. S2CID 19381209.
- ^ Sato, Kazuo; Kawasaki, Tomihisa; Taniuchi, Yuta; Hirayama, Fukushi; Koshio, Hiroyuki; Matsumoto, Yuzo (1997-11-27). "YM-60828, a novel factor Xa inhibitor: Separation of its antithrombotic effects from its prolongation of bleeding time". European Journal of Pharmacology. 339 (2–3): 141–46. doi:10.1016/S0014-2999(97)01389-7. PMID 9473127.
- ^ "Summary Basis of Decision (SBD) for PrXARELTO". Health Canada. 2009-02-13. Archived from the original on 2016-10-09. Retrieved 2016-10-03.
- ^ a b c d Nar, Herbert (2012). "The role of structural information in the discovery of direct thrombin and factor Xa inhibitors". Trends in Pharmacological Sciences. 33 (5): 279–88. doi:10.1016/j.tips.2012.03.004. PMID 22503439.
- ^ Brandstetter, Bland (1996). "X-ray Structure of Active Site-inhibited Clotting Factor Xa". The Journal of Biological Chemistry. 271 (47): 29988–92. doi:10.1074/jbc.271.47.29988. PMID 8939944.
- ^ a b c Steinberg, Benjamin A. (2014). "Structure–function relationships of factor Xa inhibitors: implications for the practicing clinician". Journal of Thrombosis and Thrombolysis. 37 (2): 234–41. doi:10.1007/s11239-013-0991-z. PMID 23996500.
- ^ Becker, Richard C.; Alexander, John; Dyke, Christopher K.; Harrington, Robert A. (2004-12-01). "Development of DX-9065a, a novel direct factor Xa antagonist, in cardiovascular disease". Thrombosis and Haemostasis. 92 (6): 1182–93. doi:10.1160/TH04-05-0289. ISSN 0340-6245. PMID 15583722. S2CID 953689.
- ^ a b Katakura, S.; Hara, T.; Nagahara, T.; Kunitada, S.; Iwamoto, M. (1995-05-01). "Molecular model of an interaction between factor Xa and DX-9065a, a novel factor Xa inhibitor: Contribution of the acetimidoylpyrrolidine moiety of the inhibitor to potency and selectivity for serine proteases". European Journal of Medicinal Chemistry. 30 (5): 387–94. doi:10.1016/0223-5234(96)88248-1.
- ^ Pinto, Orwat, Koch, Donald J.P. Michael J. Stephanie (2007). "Discovery of 1-(4-Methoxyphenyl)-7-oxo-6-(4-(2-oxopiperidin-1-yl)phenyl)-4,5,6,7-tetrahydro- 1H-pyrazolo[3,4-c]pyridine-3-carboxamide (Apixaban, BMS-562247), a Highly Potent, Selective, Efficacious, and Orally Bioavailable Inhibitor of Blood Coagulation Factor Xa". Journal of Medicinal Chemistry. 50 (22): 5339–56. doi:10.1021/jm070245n. PMID 17914785.
{{cite journal}}: CS1 maint : 복수이름 : 작성자 목록(링크) - ^ Roehrig, Susanne (2005). "Discovery of the Novel Antithrombotic Agent 5-Chloro- N -({(5 S )-2-oxo-3- [4-(3-oxomorpholin-4-yl)phenyl]-1,3-oxazolidin-5-yl". Journal of Medicinal Chemistry. 48 (19): 5900–5908. doi:10.1021/jm050101d. PMID 16161994.
- ^ a b Pinto, D. J.; Orwat, M. J.; Koch, S.; Rossi, K. A.; Alexander, R. S.; Smallwood, A.; Lam, P. Y. (2007). "Discovery of 1-(4-methoxyphenyl)-7-oxo-6-(4-(2-oxopiperidin-1-yl)phenyl)-4,5,6,7-tetrahydro-1H -pyrazolo[3,4-c]pyridine-3-carboxamide (apixaban, BMS-562247), a highly potent, selective, efficacious, and orally bioavailable inhibitor of blood coagulation factor Xa". J Med Chem. 50 (22): 5339–56. doi:10.1021/jm070245n. PMID 17914785.
- ^ Mahy, William; Leitch, Jamie A.; Frost, Christopher G. (2016-03-01). "Copper Catalyzed Assembly of N-Aryloxazolidinones: Synthesis of Linezolid, Tedizolid, and Rivaroxaban". European Journal of Organic Chemistry. 2016 (7): 1305–13. doi:10.1002/ejoc.201600033. ISSN 1099-0690.
- ^ a b c 미국 특허권 7351823, 마티아스 버웨, 크리스티안 토마스, 요아힘 레세, 더크 그로트존, 2008-04-D01 출판, 2005-01-10을 발행한 "준비 과정"
- ^ Li, Chao; Liu, Yingshuai; Zhang, Yongjun; Zhang, Xingxian (2011-07-01). "An approach to the anticoagulant agent rivaroxaban via an isocyanate-oxirane cycloaddition promoted by MgI2.etherate". Journal of Chemical Research. 35 (7): 400–01. doi:10.3184/174751911X13098778358582.
- ^ Yuan, Jianyong; Liu, Kai; Li, Lun; Yuan, Yong; Liu, Xuelei; Li, Yanwu (2014-09-18). "A Novel Synthesis of the Oxazolidinone Antithrombotic Agent Rivaroxaban". Molecules. 19 (9): 14999–15004. doi:10.3390/molecules190914999. PMC 6271174. PMID 25237754.
- ^ a b Pinto, Donald J. P.; Orwat, Michael J.; Koch, Stephanie; Rossi, Karen A.; Alexander, Richard S.; Smallwood, Angela; Wong, Pancras C.; Rendina, Alan R.; Luettgen, Joseph M. (2007-11-01). "Discovery of 1-(4-Methoxyphenyl)-7-oxo-6-(4-(2-oxopiperidin-1-yl)phenyl)-4,5,6,7-tetrahydro- 1H-pyrazolo[3,4-c]pyridine-3-carboxamide (Apixaban, BMS-562247), a Highly Potent, Selective, Efficacious, and Orally Bioavailable Inhibitor of Blood Coagulation Factor Xa". Journal of Medicinal Chemistry. 50 (22): 5339–56. doi:10.1021/jm070245n. ISSN 0022-2623. PMID 17914785.
- ^ 미국 특허 20100130543, Thomas G. Gant, Manoucherhr M.샤바즈, 2010-05-27, 2009-09-14호 발행, "인자 xa의 피라졸 카박스아미드 억제제"
- ^ Frost, C.; Song, Y.; Barret, Y .C.; Wang, J.; Pursley, J. (2014). "A randomized direct comparison of the pharmacokinetics and pharmacodynamics of apixaban and rivaroxaban". Clinical Pharmacology. 6: 179–87. doi:10.2147/CPAA.S61131. PMC 4235474. PMID 25419161.
- ^ Parasrampuriam, D.A.; Truitt, K. (2016). "Pharmacokinetics and Pharmacodynamics of Edoxaban, a Non-Vitamin K Antagonist Oral Anticoagulant that Inhibits Clotting Factor Xa". Clinical Pharmacokinetics. 55 (6): 641–55. doi:10.1007/s40262-015-0342-7. PMC 4875962. PMID 26620048.
- ^ Ahrens, I; Karlheinz, P.; Lip, GYH.; Bode, C. (June 2012). "Development and Clinical Applications of Novel Oral Anticoagulants. Part II. Drugs Under Clinical Investigation". Discovery Medicine. 13 (73): 445–50. PMID 22742650.

